Publié le 20 déc 2019Lecture 14 min
Diabètes très insulinorésistants - Focus sur les syndromes lipodystrophiques
Corinne VIGOUROUX (PU-PH), Coordinatrice du Centre de référence des pathologies rares de l’insulinosécrétion et de l’insulinosensibilité (PRISIS) ; service d’endocrinologie et laboratoire de génétique moléculaire, AP-HP, Hôpital Saint-Antoine, Paris
Les syndromes lipodystrophiques sont des maladies rares dans lesquelles un déficit du tissu adipeux conduit à des troubles métaboliques avec insulinorésistance, hypertriglycéridémie et stéatose hépatique souvent sévères. Ce sont des maladies complexes, de causes multiples, qui doivent être différenciées du syndrome métabolique « banal » pour orienter au mieux les explorations et adapter la prise en charge.
Diagnostic clinique du morphotype lipodystrophique
Le diagnostic du morphotype lipodystrophique repose sur un examen clinique soigneux à la recherche d’une perte de tissu adipeux, généralisée ou partielle.
Les syndromes lipodystrophiques se présentent sous des formes cliniques extrêmement variées, généralisées ou partielles, héréditaires ou sporadiques, présentes dès la naissance ou s’exprimant pro - gressivement à partir de l’adolescence.
Au-delà de la redistribution corporelle du tissu adipeux, qui domine le tableau clinique dans certaines formes, c’est la mise en évidence d’une perte de tissu adipeux sous-cutané, en l’absence de dénutrition, qui doit faire penser au diagnostic. La lipoatrophie s’apprécie à l’examen clinique par l’inspection et la mesure du pli cutané, que l’on peut évaluer précisément grâce à une pince de Harpenden. Au niveau des membres, la visibilité accrue des veines sous-cutanées et des reliefs musculaires secondaires à la lipoatrophie attire souvent l’attention. Lorsque la lipoatrophie atteint le visage, l’atrophie des boules de Bichat donne un aspect cachectique et l’exacerbation des reliefs osseux du visage un morphotype acromégaloïde (arcades sourci lières et pommettes saillantes) (figure 1A). La perte du tissu adipeux mécanique (palmoplantaire, articulaire) favorise les ongles incarnés et les douleurs (perte du coussinet plantaire).
Dans les formes partielles de lipoatrophie, on peut observer des zones d’accumulation du tissu adipeux. Ainsi dans le syndrome de Barraquer-Simons, le visage et le tronc sont lipo-atrophiques tandis que le tissu adipeux s’accumule à la partie inférieure du corps. À l’inverse, la lipodystrophie de Dunnigan se caractérise par une lipoatrophie des membres qui contraste avec une augmentation de l’adiposité facio-cervicale responsable d’un morphotype cushingoïde : visage rond, double menton, comblement des creux sus-claviculaires, bosse de bison. Néanmoins, d’autres signes cliniques différencient la présentation clinique du syndrome de Dunnigan de celle de l’hypercorticisme : absence de fragilité cutanée, hypertrophie musculaire souvent marquée. La lipodystrophie de Dunnigan se caractérise aussi chez les femmes par une hypomastie, une accumulation de tissu graisseux au niveau pubien avec épaississement vulvaire souvent gênant et fréquemment un hirsutisme. Le morphotype androïde (diamètre bi-acromial supérieur au diamètre bi-trochantérien), des extrémités (mains et pieds) élargies et infiltrées peuvent aussi être des motifs de consultation, en particulier chez les femmes, chez qui le diagnostic clinique est plus facile que chez les hommes. De plus, il n’y a pas d’accumulation de tissu adipeux souscutané abdominal, et l’index de masse corporelle est normal dans la majorité des cas (figure 1B). L’ensemble du tableau clinique permet en général de différencier un syndrome de Dunnigan d’un « banal » syndrome métabolique avec morphotype an droïde.
Figure 1. A. Représentation schématique d’un patient atteint de lipodystrophie généralisée. B. Représentation schématique d’une patiente atteinte de lipodystrophie partielle de Dunnigan.
Anomalies métaboliques associées à la lipodystrophie clinique
Devant une suspicion clinique de déficit partiel ou généralisé du tissu adipeux, il convient de rechercher les troubles métaboliques associés, qui permettent alors de parler de véritable syndrome lipodystrophique (figure 2).
Figure 2. Exploration des syndromes lipodystrophiques.
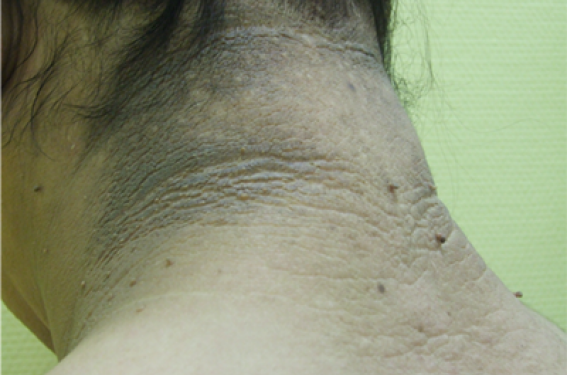
L’insulinorésistance, secondaire à la dysfonction adipocytaire, est à rechercher cliniquement sous la forme d’un acanthosis nigricans, lésion cutanée hyperkératosique et pigmentée siégeant principalement au niveau des plis corporels (axillaires, inguinaux, cervicaux). Le molluscum pendulum (ou acrochordon), aux mêmes localisations, est un équivalent sémiologique (figure 3).
Figure 3. Acanthosis nigricans et/ou molluscum pendulum (acrochordons) axillaires et/ou cervicaux chez des patients atteints de syndrome lipodystrophique.
La mesure de la glycémie et de l’insulinémie (ou du peptide C chez les patients diabétiques insulinotraités) à jeun suffit parfois à objectiver une insulinorésistance, avec une hyperinsulinémie sans diabète, une insulinosécrétion préservée et/ou la nécessité de fortes doses d’insuline chez les pa tients diabétiques, en l’absence d’anticorps spécifiques du diabète de type 1. Devant des valeurs subnormales, on pourra compléter par une hyperglycémie provoquée par voie orale, qui permet parfois de démasquer des hyperinsulinémies post-stimulatives et/ou des troubles de la tolérance au glucose. Au cours du temps apparaît néanmoins une dysfonction cellulaire bêta, et le défaut d’insulinosécrétion peut rendre l’insulinorésistance difficile à objectiver facilement au cours de l’évolution sans réaliser des épreuves métaboliques dynamiques (hyperglycémie intraveineuse, clamp euglycémique hyperinsulinémique, perfusion de glucose par paliers).
Le bilan lipidique montre le plus souvent une hypertriglycéridémie avec HDL-cholestérol bas. Ce profil caractérise les états d’insulinorésistance dus à des anomalies de la signalisation insulinique situées en aval du récepteur de l’insuline, comme c’est le cas dans les syndromes lipodystrophiques (figure 4). L’hypertriglycéridémie est parfois sévère dans les syndromes lipodystrophiques, avec un risque de pancréatite aiguë. En revanche, dans les syndromes d’insulinorésistance sévères dus aux mutations du récepteur de l’insuline, qui peuvent représenter des dia gnostics différentiels des syndromes lipodystrophiques, il n’y a généralement pas d’hypertriglycéridémie. Même si les patients atteints d’insulinorésistance due à des anomalies du récepteur de l’insuline ne présentent ni lipoatrophie caractérisée ni redistribution corporelle du tissu adipeux, faire la part entre ces diagnostics est parfois cliniquement difficile, en particulier lorsque l’hyperandrogénie est sévère et s’accompagne d’hypertrophie musculaire.
Figure 4. Le défaut de stockage de l’excédent énergétique sous forme de triglycérides adipocytaires conduit aux troubles métaboliques associés aux syndromes lipodystrophiques.
L’absorptiométrie biphotonique corps entier, qui quantifie la masse grasse totale et segmentaire, et le dosage de leptine peuvent aider à orienter le diagnostic. La leptinémie reste corrélée à la masse grasse totale dans les syndromes lipodystrophiques : effondrée dans les lipodystrophies généralisées, elle est basse en regard de l’index de masse corporelle dans les formes partielles de lipodystrophies. Les dosages d’adiponectine sériques, ainsi que de SHBG (sex-hormone steroid binding globulin), bas dans les lipodystrophies, mais normaux, voire élevés dans les altérations primitives du récepteur insulinique, peuvent aussi aider à différencier les syndromes lipodystrophiques des syndromes d’insulinorésistance liés aux dysfonctions primitives du récepteur de l’insuline.
L’hirsutisme est fréquent chez les femmes lipodystrophiques, en rapport avec un syndrome des ovaires polykystiques qui évolue de façon parallèle à l’insulinorésistance. Il n’est pas rare qu’il soit le mode de révélation de la maladie. L’examen clinique, la recherche d’une hyperandrogénie d’origine ovarienne et l’évaluation morphologique des ovaires font partie des examens utiles chez les femmes atteintes.
La stéatose hépatique est un signe précoce, qui résulte à la fois de l’hyperinsulinémie et du stockage ectopique des lipides. Elle peut être au premier plan du tableau clinique, en particulier en période néonatale chez les enfants atteints de lipodystrophie généralisée congénitale. Elle devra faire l’objet d’une surveillance attentive, car elle peut évoluer vers la cytolyse, la cirrhose avec hypertension portale, l’insuffisance hépatique.
Interrogatoire
L’interrogatoire est très important, non seulement pour retracer l’évolution de la maladie, mais aussi pour rechercher des antécédents familiaux ou une consanguinité parentale.
Un point majeur de l’exploration des patients est l’interrogatoire, et l’observation des photographies antérieures. Les lipodystrophies généralisées congénitales peuvent être dia gnostiquées dans les premiers mois de vie devant une lipoatrophie néonatale quasi complète. Néanmoins, si les troubles métaboliques (insulinorésistance, hypertriglycéridémie, stéatose hépatique) sont précoces, ils peuvent être d’évolution spontanément favorable dans la petite enfance et ne réapparaître qu’en période pubertaire ou post-pubertaire, avec en particulier un diabète qui ne se développe en général qu’à l’adolescence. Le diagnostic de lipoatrophie généralisée congénitale peut donc tout à fait être fait par les médecins d’adultes. Les syndromes lipodystrophiques partiels, même d’origine génétique, ne s’expriment en général cliniquement qu’à partir de la période pubertaire.
L’interrogatoire recherche aussi des antécédents familiaux, non seulement de lipodystrophie, souvent sous-diagnostiquée, mais aussi de diabète atypique (précoce, sévère, non clairement étiqueté, chez un sujet non obèse), de dyslipidémie sévère, d’hypertrophie musculaire spontanée, d’hyperandrogénie chez les femmes, qui orientent vers une forme autosomale dominante de la maladie. Il est également très important d’interroger les patients sur la possibilité d’une consanguinité parentale, soit connue, soit envisagée, par exemple en cas d’endogamie (parents originaires de villages voisins), qui donnerait alors un argument pour une forme autosomale récessive de syndrome lipodystrophique.
La mise en évidence de variants génétiques spécifiques permet d’affirmer le diagnostic
La connaissance des gènes responsables des syndromes lipodystrophiques a permis de révéler l’importance des capacités de stockage énergétique et des fonctions endocrines du tissu adipeux dans l’homéostasie métabolique systémique (tableaux 1 et 2). La liste des gènes responsables de syndromes lipodystrophiques s’allonge régulièrement, et on connaît aujourd’hui plus de 25 gènes différents dont les variants pathogènes sont responsables de formes monogéniques de syndromes lipodystrophiques. Devant un syndrome lipodystrophique, le diagnostic étiologique le plus efficace passe donc aujourd’hui, comme nous le proposons au laboratoire de génétique moléculaire de l’hôpital Saint-Antoine à Paris, par le séquençage d’un panel de gènes spécifiques, remis à jour régulièrement, permettant d’explorer au mieux l’hypothèse d’une forme monogénique. Ce diagnostic est important, car il permet d’orienter au mieux à la fois les explorations complémentaires et le traitement. Une fois le diagnostic affirmé chez le cas index, le dépistage familial permettra de prendre en charge le plus tôt possible les sujets atteints, et d’éviter que le diagnostic ne soit fait dans des circonstances parfois dramatiques (pancréatite aiguë révélant une hypertriglycéridémie méconnue par exemple).
Formes étiologiques particulières
Certains contextes pathologiques (atteintes spécifiques d’organe, maladies auto-immunes ou autoinflammatoires associées, antécédents évocateurs) orientent vers des formes étiologiques particulières de syndromes lipodystrophiques.
Les syndromes lipodystrophiques sont pour la plupart associés non seulement aux troubles métaboliques décrits plus hauts, mais aussi à d’autres manifestations qui témoignent d’une atteinte multitissulaire. Ainsi, les lipodystrophies généralisées congénitales s’associent souvent à des atteintes osseuses, principalement sous la forme de kystes ostéolytiques, en général asymptomatiques, qui peuvent orienter le diagnostic. Certaines formes de laminopathies, des maladies liées aux mutations des lamines A/C, des protéines nucléaires structurant le noyau cellulaire, peuvent s’exprimer sous la forme de syndromes de vieillissement accéléré comportant, parmi d’autres signes (entre autres dysmorphie faciale, anomalies chondro-osseuses, alopécie, troubles trophiques des extrémités, rétractions tendineuses, calcifications artérielles et valvulaires), un syndrome lipodystrophique généralisé ou partiel. Dans certaines formes génétiques de lipomatose de Launois-Bensaude coexistent des masses adipeuses souvent volumineuses prédominant aux racines des membres et une lipoatrophie marquée des zones non lipomateuses avec leptinémie basse et altérations métaboliques associées à l’insulinorésistance. Une neuropathie périphérique, souvent méconnue, est fréquente dans cette forme particulière de syndrome lipodystrophique, ainsi que dans d’autres formes étiologiques. Des cardiopathies, des atteintes digestives, des anomalies neuro-sensorielles s’associent à certains syndromes lipodystrophiques. Des syndromes auto-inflammatoires systémiques, résultant d’altérations génétiques de l’immunoprotéasome, comportent parfois une lipoatrophie sévère avec fièvre et panniculite.
Les syndromes lipodystrophiques n’ont pas tous une origine génétique connue. C’est le cas des formes associées aux maladies dysimmunitaires, soit systémiques (lupus, sclérodermie, etc.), soit témoignant d’une atteinte autoimmune d’organe (thyroïdite, hépatite auto-immune, etc.), que l’on regroupe sous le terme de lipodystrophies « acquises. Parmi celles-ci, le syndrome de Barraquer-Simons associe une lipoatrophie de la partie supérieure du corps et une accumulation de tissu adipeux à la partie inférieure, avec dans 20 % des cas environ une glomérulopathie avec protéinurie et anomalies du complément. Des syndromes lipodystrophiques ont également été rapportés à distance d’irradiations corporelles totales et de greffes de moelle chez des patients aux antécédents d’hémopathies malignes.
L’agression immuno-inflammatoire du tissu adipeux pourrait donc conduire à de véritables syndromes lipodystrophiques.
Enfin, les traitements antirétroviraux de l’infection par le VIH ont été d’importants pourvoyeurs de syndromes lipodystrophiques avec insulinorésistance et troubles métaboliques associés. Les traitements actuels sont moins toxiques pour le tissu adipeux, mais certains patients gardent des séquelles définitives des traitements antirétroviraux initiaux.
Le diagnostic de syndrome lipodystrophique implique de rechercher, selon la cause, des comorbidités spécifiques qui pourront bénéficier d’une prise en charge spécifique.
Chaque forme étiologique différente de syndrome lipodystrophique est associée à des comorbidités spécifiques, qui sont très diverses et ne seront pas listées en détail ici. L’exploration initiale des patients pourra être faite en milieu spécialisé. À cet égard, l’expertise du centre de référence PRISIS (Pathologies rares de l’insulinosécrétion et de l’insulinosensibilité) et de son réseau de centres de compétence sur l’ensemble du territoire français pourra être utile.
Dans le syndrome de Dunnigan, le moins rare des syndromes lipodystrophiques d’origine génétique, il est nécessaire de rechercher des atteintes cardiovasculaires associées, sous la forme de cardiopathies avec troubles du rythme et de la conduction cardiaques nécessitant au minimum une échographie et un holter cardiaque sur 24 h, et/ou d’athérosclérose, en particulier coronarienne, qui peut être particulièrement précoce et sévère et nécessite des examens de dépistage systématiques (score calcique, test d’effort).
Prise en charge
Le traitement des troubles métaboliques nécessite en premier lieu l’instauration de règles hygiéno-diététiques privilégiant l’activité physique régulière et évitant tout apport énergétique excédentaire.
Les syndromes lipodystrophiques sont des maladies difficiles à traiter. Aucune intervention thérapeutique actuelle n’est capable de restaurer la fonctionnalité du tissu adipeux dans les zones lipoatrophiques et la prise en charge de l’insulinorésistance est souvent décevante. Les règles hygiéno-diététiques doivent être instaurées le plus précocement possible, dès le diagnostic, même en l’absence de trouble métabolique patent. Il est nécessaire d’obtenir un bon équilibre alimentaire et d’éviter tout apport énergétique excédentaire. Chez le nourrisson l’utilisation des triglycérides à chaîne moyenne est souvent efficace pour limiter l’hypertriglycéridémie. L’activité physique est capable d’améliorer significativement l’insulinosensibilité, et les patients doivent y être sensibilisés. En première intention, le traitement du diabète, de la dyslipidémie, et des complications associées utilise l’arsenal thérapeutique non spécifique. La metformine est utilisée précocement ; les agonistes des récepteurs du GLP1 sont utiles pour traiter l’hyperglycémie et réduire l’hyperphagie qui s’associe au déficit en leptine ; l’insuline nécessite souvent d’être administrée sous forme ultra-concentrée ; les fibrates sont utilisés en cas d’hypertriglycéridémie importante. Les patients, même avant qu’ils ne développent un diabète, doivent être considérés comme à haut risque cardiovasculaire et les statines sont souvent nécessaires pour maintenir un LDL-cholestérol bas. Les estrogènes de synthèse sont contreindiqués en raison du risque de poussée d’hypertriglycéridémie. Un suivi cardiovasculaire doit être systématiquement instauré, et la prise en charge multidisciplinaire doit être adaptée aux comorbidités spécifiques à chaque forme étiologique.
Le morphotype stigmatisant des patients peut être la source d’une souffrance psychologique importante. L’utilisation de produits de comblement (injections au niveau des joues en particulier) et/ou la chirurgie plastique (liposuccion, résection chirurgicale de tissu adipeux, injection de tissu adipeux autologue) peuvent être utiles.
L’association de patients AFLIP (Association française des lipodystrophies) peut aider les patients et leur famille.
Le traitement substitutif par la metreleptine peut être utile dans certaines formes sévères de syndromes lipodystrophiques.
L’utilisation de la leptine recombinante (metreleptine), dans le contexte du déficit en leptine secondaire à la lipoatrophie, n’a pas fait l’objet d’études contre placebo. Néanmoins, même si la metreleptine n’améliore pas la lipoatrophie, il a été montré qu’elle s’oppose au stockage ectopique des graisses, à la fois en diminuant l’hyperphagie, souvent difficile à gérer, des patients très déficients en leptine et en activant des voies insulinosensibilisatrices indépendantes de la prise alimentaire. Son efficacité sur les troubles métaboliques associés aux formes généralisées de lipoatrophie a également été bien documentée.
La metreleptine permet d’améliorer la sensibilité à l’insuline et de faire régresser l’hypertriglycéridémie, l’hyperglycémie, et la stéatose hépatique chez la plupart des patients atteints de lipoatrophie généralisée. Elle semble avoir une efficacité moindre dans les formes partielles de lipodystrophies, et doit être réservée aux formes avec troubles métaboliques sévères et déficit majeur en leptine (tableau 1).
La metreleptine a obtenu une AMM européenne en 2018 dans le traitement des complications métaboliques associées au déficit en leptine chez les patients lipodystrophiques, en complément d’un régime alimentaire, à partir de l’âge de 2 ans dans les lipoatrophies généralisées congénitales, et à partir de 12 ans dans les formes partielles de lipodystrophie lorsque les traitements standard n’ont pas permis d’obtenir un contrôle métabolique suffisant. Un avis favorable pour ces indications a été rendu par la commission de transparence de la HAS en février 2019, qui a souhaité que cette option thérapeutique soit validée et régulièrement réévaluée au cours des réunions de concertation pluridisciplinaires du centre de référence Maladies rares PRISIS, et que les patients traités consultent au moins une fois par an dans le centre de référence ou les centres de compétence du réseau PRISIS.
La metreleptine s’administre en une injection sous-cutanée quotidienne. Les patients doivent apprendre à préparer la solution injectable et à doser correctement le produit dans une seringue à remplir extemporanément, ce qui peut limiter l’observance thérapeutique. Les doses utilisées, jusqu’à 10 mg/j (1 flacon par jour), sont adaptées aux réponses métaboliques et à la tolérance. Les principaux effets secondaires sont les réactions locales aux sites d’injection et les hypoglycémies en début de traitement, nécessitant d’anticiper la diminution des doses d’insuline. La metreleptine entraîne habituellement une perte de poids, qui fait partie de l’effet thérapeutique. Le développement d’auto-anticorps anti-leptine est fréquent, mais ils ne sont qu’exceptionnellement neutralisants. De très rares cas de lymphomes ont été décrits chez des patients atteints de lipodystrophie généralisée acquise sous traitement par metreleptine. La pathologie dysimmunitaire sous-jacente, responsables d’anomalies hématologiques préexistant au traitement dans 2 des 3 cas décrits, a été considérée comme un facteur de risque important, mais le rôle exact de la metreleptine sur la croissance tumorale reste inconnu. Un registre de surveillance post-AMM est prévu. Le prix et les conditions de remboursement de la metreleptine sont en cours d’évaluation par la HAS en France.
Conclusion
Les syndromes lipodystrophiques sont des maladies rares associant déficit en tissu adipeux et troubles métaboliques avec insulinorésistance.
L’examen clinique et la recherche des anomalies métaboliques sont à la base du diagnostic.
Le diagnostic génétique permet d’orienter la recherche de comorbidités spécifiques, d’améliorer la prise en charge, et de dépister et traiter le plus tôt possible les apparentés atteints.
Un suivi multidisciplinaire est nécessaire. La prise en charge des complications métaboliques nécessite en premier lieu l’instauration précoce de règles hygiéno-diététiques privilégiant l’exercice physique et évitant tout apport énergétique excédentaire.
L’utilisation de la metreleptine doit être réservée aux syndromes lipodystrophiques sévères en échec thérapeutique.
Le réseau des centres de référence et de compétence Maladies rares PRISIS propose une aide au diagnostic et à la prise en charge des patients atteints de syndromes lipodystrophiques.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :