Physiologie-Physiopathologie
Publié le 31 aoû 2006Lecture 15 min
Physiopathologie du diabète de type 2 - Rôles respectifs de l’insulinorésistance et du déficit en insuline
J. GIRARD, Institut Cochin, CNRS UMR 8104, INSERM U 567, Université Paris V, Paris
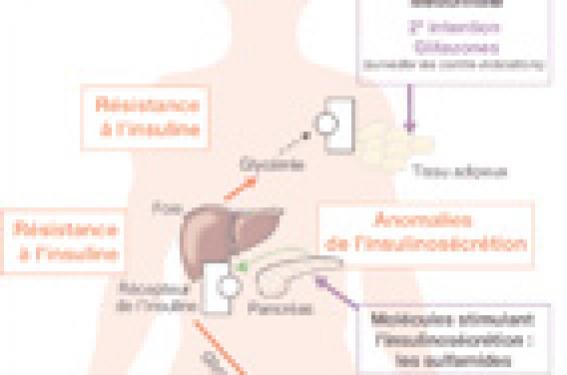
Le diabète de type 2 apparaît lorsque coexistent deux anomalies majeures : une réduction (insulinorésistance) des effets de l’insuline sur ses tissus cibles (foie et muscles squelettiques), et une diminution quantitative et qualitative (phase précoce, pulsatilité) de la sécrétion d’insuline (figure 1). Le diabète de type 2 est aussi associé à une augmentation de la sécrétion de glucagon, qui est souvent négligée, mais qui a des conséquences importantes sur la production hépatique de glucose. L’insulinorésistance se caractérise par une surproduction de glucose par le foie et une réduction de l’utilisation du glucose par les muscles squelettiques. Elle résulte d’un défaut de la voie de signalisation de l’insuline, secondaire au dysfonctionnement du tissu adipeux. Le diabète de type 2 n’apparaît jamais tant que les cellules β du pancréas sont capables de compenser l’insulinorésistance par une sécrétion accrue d’insuline (état prédiabétique).
Le passage de l’état prédiabétique au diabète de type 2 se caractérise par trois changements :
- le premier est une diminution de la fonction des cellules β du pancréas et de l’insulinosécrétion compensatrice. On ne sait pas si cette perte de fonction est génétiquement programmée ou acquise (glucotoxicité ou lipotoxicité) ou les deux à la fois. Cette transition est cruciale dans l’histoire naturelle du diabète de type 2 ;
- le second est une augmentation de la production hépatique de glucose, probablement secondaire à la sécrétion excessive de glucagon, à l’excès d’acides gras libres et d’adipocytokines libérés par le tissu adipeux ;
- le troisième est une augmentation de la résistance des muscles à l’insuline, souvent liée à la présence d’une obésité et d’un excès d’acides gras libres et d’adipocytokines.
L’objectif de cet article est de résumer nos connaissances des mécanismes biochimiques responsables des anomalies de la sécrétion des hormones pancréatiques et de l’action de l’insuline, et d’essayer d’identifier les étapes cellulaires qui devraient être à la base des approches pharmacologiques du traitement du diabète de type 2.
Mécanismes de la résistance musculaire et hépatique à l’insuline
L’utilisation de techniques variées, principalement la technique du clamp euglycémique hyperinsulinémique, a permis de mettre en évidence un état de résistance à l’insuline chez les diabétiques de type 2. Comme 80 % des diabétiques de type 2 sont obèses et que l’obésité est un facteur de résistance à l’insuline, il est important de distinguer ce qui est dû au diabète de ce qui est dû à l’obésité. La résistance à l’insuline est observée chez les sujets diabétiques minces comparés à des témoins minces, et les sujets diabétiques obèses sont plus résistants à l’insuline que des témoins ayant le même degré d’obésité, si bien que l’insulinorésistance des sujets diabétiques est spécifique de l’état diabétique. Par souci de simplicité, nous ne parlerons, dans la suite de ce texte, que de diabète de type 2, même lorsque le diabète est associé à une obésité.
Chez les sujets diabétiques, la production hépatique de glucose est moins freinée et l’utilisation périphérique de glucose moins stimulée en réponse à l’insuline, indiquant que le foie et les tissus périphériques sont impliqués dans l’insulinorésistance. Chez l’homme, les muscles squelettiques sont le site majeur des effets de l’insuline sur le captage périphérique de glucose.
Il a été rapidement établi que l’insulinorésistance des muscles et du foie ne résulte pas de la diminution du nombre de récepteurs de l’insuline mais d’un défaut d’activation de la voie de signalisation de l’insuline (figure 1).
Figure 1. Représentation schématique des anomalies de l’homéostasie du glucose chez les diabétiques de type 2. Le diabète de type 2 résulte de 3 anomalies : 1) une diminution de l’utilisation de glucose dans les muscles squelettiques, 2) une hyperproduction de glucose par le foie, 3) une sécrétion réduite d’insuline et une sécrétion exacerbée de glucagon par le pancréas.
De nouvelles techniques, basées sur la résonance magnétique nucléaire, ont permis d’identifier in vivo les étapes cellulaires défectueuses dans le muscle des diabétiques de type 2. Elles ont montré que l’insulinorésistance musculaire résulte d’un défaut de stimulation de la synthèse musculaire de glucose en réponse à l’insuline et que ce défaut résulte lui-même d’une diminution du transport de glucose en réponse à l’insuline. Ce défaut semble résulter d’une inhibition de la phosphorylation sur tyrosine du substrat du récepteur de l’insuline (IRS), conduisant à une diminution de l’association d’IRS à la phosphatidylinositol 3 kinase (PI 3 kinase) et à une diminution de la cascade de signalisation conduisant à la stimulation du transport de glucose (figure 2). Le transport de glucose est l’étape limitante du métabolisme du glucose dans le muscle. La stimulation du transport de glucose par l’insuline résulte d’une translocation de transporteurs spécifiques de glucose (Glut4) localisés dans les membranes intracellulaires vers la membrane plasmique. La stimulation du transport de glucose par l’insuline est diminuée de 50 % dans le muscle des diabétiques de type 2 et elle est corrélée à la sévérité du diabète. Comme la concentration de Glut4 dans les muscles des sujets diabétiques est peu altérée, un défaut dans la translocation (ou le trafic) de Glut4 est à l’origine de la diminution du transport de glucose.
Figure 2. La voie de signalisation de l’insuline et la stimulation du transport de glucose. Une fois liée à la sous-unité alpha de son récepteur, l’insuline induit un changement de conformation de la sous-unité β intracellulaire. Celle-ci est autophosphorylée sur des résidus tyrosine (Tyr-P) et acquiert une activité tyrosine kinase qui lui permet de phosphoryler sur des résidus tyrosine (Tyr-P) son principal substrat intracellulaire, l’IRS (Insulin Receptor Substrate). L’IRS phosphorylé recrute alors d’autres protéines pour stimuler le transport du glucose. La liaison de l’IRS phosphorylé sur tyrosines à la sous-unité régulatrice (p85) de la phosphatidylinositol 3-kinase (PI 3-K), conduit à son activation. La sous-unité catalytique de la PI 3-K (p110) permet la phosphorylation de phosphatidylinositols en position 3, et l’accumulation de PI 3. Ces molécules stimulent ensuite des sérine/thréonine kinases (par exemple la protéine kinase B), ce qui aboutit à la translocation des transporteurs de glucose Glut4, de la vésicule intracellulaires où ils sont stockés, vers la membrane plasmique.
Mécanismes du défaut de signalisation insulinique
L’un des mécanismes qui explique cette diminution de la signalisation insulinique est l’accumulation intracellulaire de métabolites lipidiques (acyl-CoA, diacylglycérol) dans le muscle squelettique et dans le foie, résultant de l’excès de libération d’acides gras par le tissu adipeux. En effet, le tissu adipeux des diabétiques de type 2 étant moins sensible à l’insuline, la lipolyse n’est pas inhibée et le tissu adipeux libère des quantités considérables d’acides gras. L’accumulation d’acyl-CoA et de diacylglycérol stimule une protéine kinase C qui phosphoryle IRS sur des résidus sérine, ce qui conduit à la diminution de la cascade de signalisation insulinique (figure 3).
L’autre mécanisme est la libération accrue par le tissu adipeux des diabétiques de type 2 obèses d’adipocytokines (TNF-α, interleukine-6, résistine) inhibant la voie de signalisation insulinique. La diminution de la sécrétion d’une adipocytokine insulinosensibilisatrice (l’adiponectine) par le tissu adipeux des diabétiques de type 2 obèses contribue également à l’insulinorésistance. En effet, les études réalisées in vivo et in vitro chez les rongeurs ont clairement démontré que l’adiponectine stimule le transport de glucose (muscle) et l’oxydation des acides gras (muscle et foie) via l’activation d’une protéine kinase dépendante de l’état énergétique de la cellule (AMP kinase) (figure 4).
Figure 3. Effets des acides gras sur la voie de signalisation de l’insuline et la stimulation du transport de glucose.
Figure 4. Mécanisme d’action de l’adiponectine sur le transport de glucose.
Mécanismes de l’hyperproduction hépatique de glucose
Les diabétiques de type 2 ont une production hépatique de glucose augmentée en période postabsorptive, et il existe une étroite corrélation entre le degré de l’hyperglycémie et la production hépatique de glucose. L’hyperproduction de glucose par le foie des sujets diabétiques résulte d’une augmentation de la gluconéogenèse, la glycogénolyse n’étant pas augmentée. Deux facteurs peuvent contribuer à l’augmentation de la gluconéogenèse par le foie des sujets diabétiques.
Le premier pourrait être l’existence d’une hyperglucagonémie tout au long de la journée chez les sujets diabétiques, qui stimule la gluconéogenèse secondairement à l’augmentation de l’expression de gènes codant des enzymes clés de cette voie métabolique, comme la phosphoénolpyruvate carboxykinase (PEPCK) et la glucose-6-phosphatase (G-6-Pase). La suppression de l’hyperglucagonémie réduit l’hyperproduction de glucose par le foie chez les sujets diabétiques et la surexpression de la PEPCK dans le foie de souris transgénique conduit à un état d’intolérance au glucose, mais pas au diabète non insulinodépendant. Cela suggère que l’hyperproduction de glucose par le foie contribue au développement de l’intolérance au glucose, mais ne peut en aucun cas être responsable à elle seule du diabète de type 2.
Le second facteur pourrait être la présence de concentrations élevées d’acides gras libres tout au long de la journée chez les sujets diabétiques, dont l’oxydation hépatique fournissant les cofacteurs (ATP, acétyl-CoA, NADH) indispensables au bon fonctionnement de la gluconéogenèse. L’inhibition de l’oxydation hépatique des acides gras conduit, en effet, à une diminution de la production hépatique de glucose chez les sujets diabétiques.
Enfin, la gluconéogenèse étant moins sensible que la glycogénolyse à l’action inhibitrice de l’insuline, la présence d’une gluconéogenèse active chez les sujets diabétiques pourrait expliquer la moindre efficacité de l’insuline sur le foie.
En période postabsorptive, ou à la suite d’un test de tolérance au glucose administré par voie orale, la production hépatique de glucose (glycogénolyse et gluconéogenèse) n’est pas freinée chez les sujets diabétiques, contrairement à ce qui est observé chez les non-diabétiques. Comme l’absorption intestinale de glucose et l’utilisation périphérique de glucose (en raison de l’hyperglycémie) des sujets diabétiques sont semblables à celles mesurées chez les non-diabétiques, l’absence d’inhibition de la production hépatique de glucose est responsable de l’hyperglycémie postabsorptive.
Plusieurs facteurs pourraient contribuer à ce phénomène :
– la moindre suppression de la sécrétion du glucagon et de la lipolyse et la sécrétion retardée d’insuline ;
– l’absence d’inhibition de la production hépatique de glucose en réponse à l’hyperglycémie chez les sujets diabétiques.
Anomalies de la sécrétion du pancréas exocrine
Les anomalies de la sécrétion d’insuline
Des études longitudinales réalisées chez le même sujet à différents stades du diabète ont montré une détérioration de l’insulinosécrétion (figure 5) au fur et à mesure de l’évolution de la maladie. Le diabète de type 2 se caractérise par :
– une perte de la phase précoce de la sécrétion insulinique en réponse au glucose, alors que la phase précoce de la sécrétion insulinique en réponse à l’arginine, au glucagon, aux hormones gastro-intestinales, à l’isoprotérénol et aux sulfonylurées est normale ou très proche de la normale ;
– la disparition de l’effet potentialisateur du glucose sur les autres sécrétagogues ;
– une abolition de la pulsatilité sécrétoire de l’insuline ;
– un pourcentage plus élevé de pro-insuline dans le plasma, suggérant un défaut de clivage de la pro-insuline en insuline dans les cellules β du pancréas.
Figure 5. Sécrétion d’insuline par la cellule β du pancréas. Sécrétion d’insuline en réponse au glucose par la cellule β des îlots de Langerhans du pancréas endocrine. Le glucose entre dans la cellule β par un transporteur spécifique de glucose GLUT2, qui équilibre rapidement la concentration de glucose intracellulaire avec celle du milieu extérieur. Le glucose est ensuite phosphorylé en glucose-6-phosphate par la glucokinase, une hexokinase à haut Km qui contrôle le flux glycolytique et génère de l’ATP. L’augmentation du rapport ATP/ADP conduit à la fermeture du canal potassique ATP-dépendant, puis à une dépolarisation membranaire et à l’ouverture du canal calcique voltage-dépendant. L’augmentation du calcium dans le cytosol stimule l’exocytose des granules d’insuline.
Le défaut de la sécrétion d’insuline chez les sujets diabétiques est lié à une mauvaise reconnaissance du glucose comme signal direct et comme agent potentialisateur de l’insulinosécrétion. Ce défaut de reconnaissance du glucose par les cellules β du pancréas des sujets diabétiques peut résulter d’une lésion intrinsèque (génétique) des cellules β et/ou de facteurs circulants (glucose, catécholamines via leurs récepteurs β2-adrénergiques) ou locaux (somatostatine, galanine, prostaglandines) exerçant des effets inhibiteurs. L’origine du défaut fonctionnel de la cellule β, génétique ou acquise, n’est pas établie. Comme le défaut de la pulsatilité de l’insulinosécrétion est présent chez les sujets diabétiques avant l’apparition de l’intolérance au glucose ou d’une hyperglycémie franche, une lésion génétique initiale des cellules β pancréatiques n’est pas exclue.
L’abolition de la pulsatilité sécrétoire de l’insuline chez les sujets diabétiques a des conséquences directes sur la sensibilité à l’insuline des tissus périphériques. En effet, l’insuline administrée de façon pulsatile est plus efficace sur la stimulation de l’utilisation de glucose que l’insuline administrée de façon continue. La nature pulsatile de l’insulinosécrétion résulte, en effet, d’oscillations de la concentration du calcium intracellulaire en réponse aux changements du métabolisme du glucose dans la cellule β. Il faut donc rechercher un défaut génétique éventuel des protéines qui contrôlent le métabolisme du glucose et/ou des canaux ioniques impliqués dans la régulation de la concentration du calcium intracellulaire.
On sait qu’une forme particulière de diabète de type 2, le MODY (Maturity-Onset Diabetes of the Young) résulte de mutations dans des gènes contrôlant le développement, le transport et le métabolisme du glucose dans les cellules β du pancréas. Néanmoins, ces mutations sont beaucoup plus rares chez les sujets diabétiques de la maturité et on peut se demander si ces anomalies peuvent expliquer les défauts de la sécrétion d’insuline chez les sujets diabétiques.
L’hypothèse qui prévaut en ce qui concerne le défaut acquis de l’insulinosécrétion est celle appelée incorrectement « glucotoxicité ». Selon cette hypothèse, l’hyperglycémie chronique aurait des effets inhibiteurs sur la sécrétion d’insuline en réponse au glucose. La phase précoce de l’insulinosécrétion résulte d’une augmentation du métabolisme du glucose qui, en augmentant l’ATP intracellulaire, conduit à la fermeture des canaux potassiques sensibles à l’ATP, à la dépolarisation de la membrane plasmique et à l’ouverture des canaux calciques dépendants du potentiel de membrane. L’augmentation du calcium intracellulaire induit alors la sécrétion d’insuline par exocytose (figure 5).
Les mécanismes intracellulaires invoqués pour expliquer le défaut de l’insulinosécrétion chez les sujets diabétiques sont nombreux : diminution du nombre de transporteurs de glucose, diminution de l’activité de la glucokinase, accumulation de glycogène, etc., mais aucun ne permet seul d’apporter une réponse définitive.
Une autre hypothèse a été récemment suggérée pour les sujets diabétiques obèses : l’accumulation de triglycérides dans le pancréas, secondairement à l’augmentation chronique de la concentration des acides gras et des triglycérides dans le plasma, conduirait à une « lipotoxicité » entraînant la perte de l’insulinosécrétion en réponse au glucose. L’accumulation de triglycérides dans les cellules β entraînerait la formation de monoxyde d’azote et de céramide, molécules qui conduiraient à une apoptose (mort cellulaire programmée) des cellules β (figure 6).
Chez les sujets diabétiques, les effets inhibiteurs de l’hyperglycémie sur l’insulinosécrétion pourraient dépendre d’une hypersensibilité à un tonus alpha-adrénergique inhibiteur ou à une production augmentée d’opioïdes ou de prostaglandines. En effet, la phase précoce de l’insulinosécrétion en réponse au glucose est partiellement restaurée par l’administration d’antagonistes β2-adrénergiques, de naloxone (un antagoniste des opioïdes endogènes) et d’inhibiteurs non stéroïdiens de la synthèse des prostaglandines PGE2 (type aspirine), même si les sujets diabétiques sont hyperglycémiques.
Figure 6. Effets provoqués par la présence d’une augmentation chronique de la concentration des acides gras libres. Les acides gras libres peuvent provenir soit de la lipolyse à partir des triglycérides du tissu adipeux, soit de la mobilisation des triglycérides du muscle ou du foie.
Les anomalies de la sécrétion du glucagon
Les sujets diabétiques ont une hyperglucagonémie relative qui reflète une anomalie de la régulation de la fonction des cellules β du pancréas. La sécrétion de glucagon est anormale chez les sujets diabétiques. La sécrétion de glucagon en réponse à l’arginine, à un repas riche en protéines ou à l’hypoglycémie insulinique est supérieure à la normale. L’inhibition de la sécrétion de glucagon en réponse à l’hyperglycémie est inférieure à la normale. Le même type d’anomalie existe chez les sujets prédiabétiques qui ont une intolérance au glucose. Cela suggère que l’altération de la sécrétion de glucagon est une lésion précoce du pancréas endocrine, qui précède l’apparition du diabète de type 2.
Le mauvais fonctionnement des cellules β semble résulter d’un défaut intrinsèque de la reconnaissance du glucose (cécité) due à l’hyperglycémie chronique (glucotoxicité). En effet, l’hyperglucagonémie et la « cécité » au glucose des cellules β sont corrigées après normalisation de la glycémie. Les mécanismes cellulaires responsables de la « cécité » au glucose des cellules β demeurent inconnus.
Le diabète de type 2 est une pathologie hétérogène, et il serait naïf de vouloir l’identifier à une maladie monogénique. Il existe probablement, chez un nombre très réduit (< 5 %) de sujets diabétiques, des défauts portant sur un seul gène (glucokinase, glycogène synthase, ADN mitochondrial, récepteur de l’insuline) mais, chez la grande majorité des sujets diabétiques, la maladie résulte d’anomalies de plusieurs gènes et des dérèglements métaboliques à long terme (hyperglycémie, hyperlipidémie). Cela est bien illustré par les expériences réalisées sur les souris transgéniques. La surexpression des enzymes clés de la néoglucogenèse dans le foie, la réduction de la glucokinase dans les cellules β du pancréas, la surexpression d’un récepteur de l’insuline déficient en activité tyrosine kinase dans les muscles et l’invalidation du transporteur de glucose GLUT4 dans les muscles et le tissu adipeux conduisent à un état d’intolérance au glucose, mais jamais à un diabète. Cela est en accord avec les études sur l’étiologie du diabète de type 2, qui suggèrent que le développement du diabète nécessite, à la fois, un défaut génétique dans les tissus cibles de l’insuline et une incapacité des cellules β à compenser ce défaut. Une partie des anomalies de la sécrétion de l’insuline et des effets de l’insuline sur ses tissus cibles semble résulter de dérèglements métaboliques à long terme (hyperglycémie, hyperlipidémie). Ceci est bien illustré par le fait que les anomalies de la sécrétion de l’insuline et des effets de l’insuline sur ses tissus cibles sont corrigées par un contrôle strict de la glycémie (perte de poids, insulinothérapie, drogues antidiabétiques, etc.).
L’identification des étapes biochimiques, dont le fonctionnement est altéré dans le diabète de type 2, est d’une importance cruciale pour permettre à l’industrie pharmaceutique de développer des substances ayant pour cibles les enzymes ou protéines impliquées dans ces défauts. On peut concevoir, en effet, que la mise au point de molécules permettant d’augmenter spécifiquement le flux de glucose (en agissant sur la glucokinase) et/ou les flux calciques (en agissant sur le canal calcique membranaire ou les calciosomes intracellulaires) dans la cellule β aurait un impact considérable sur la sécrétion d’insuline. De même, la mise au point de molécules permettant d’augmenter spécifiquement l’activité de la glycogène synthase ou d’inhiber de façon spécifique la sécrétion de glucagon aurait un intérêt considérable pour améliorer la sensibilité des tissus périphériques à l’insuline et pour freiner la gluconéogenèse chez les sujets diabétiques.
Cibles d’action des antidiabétiques oraux.
Conclusions
Les études réalisées ces dernières années ont permis d’identifier un certain nombre de mécanismes susceptibles d’expliquer les défauts de l’insulinosécrétion et de l’action de l’insuline chez les diabétiques de type 2.
Les défauts de la pulsatilité de la sécrétion d’insuline et de la sécrétion précoce d’insuline en réponse au glucose semblent être en partie liés à des anomalies du fonctionnement de la glucokinase et des canaux ioniques.
La résistance à l’insuline des diabétiques de type 2 est liée à une anomalie de la voie de signalisation de l’insuline, secondairement à l’apport accru d’acides gras et d’adipocytokines (sujets diabétiques obèses).
Enfin, l’hyperproduction de glucose par le foie résulte d’une augmentation de la gluconéogenèse, liée principalement à l’hyperglucagonémie et secondairement à l’apport accru d’acides gras (sujets diabétiques obèses).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



